











RANKIN BIOMEDICAL
HISTOLOGY SUPPLY SPECIALISTS
Since 1995, Rankin Biomedical has served the anatomic pathology industry, guided by our core values that drive our daily decisions: Put People First, Find Solutions, Obsess Over Excellence, Lead Through Virtue
Solutions
Shop our products online
Rankin distributes many of the top histology brands.
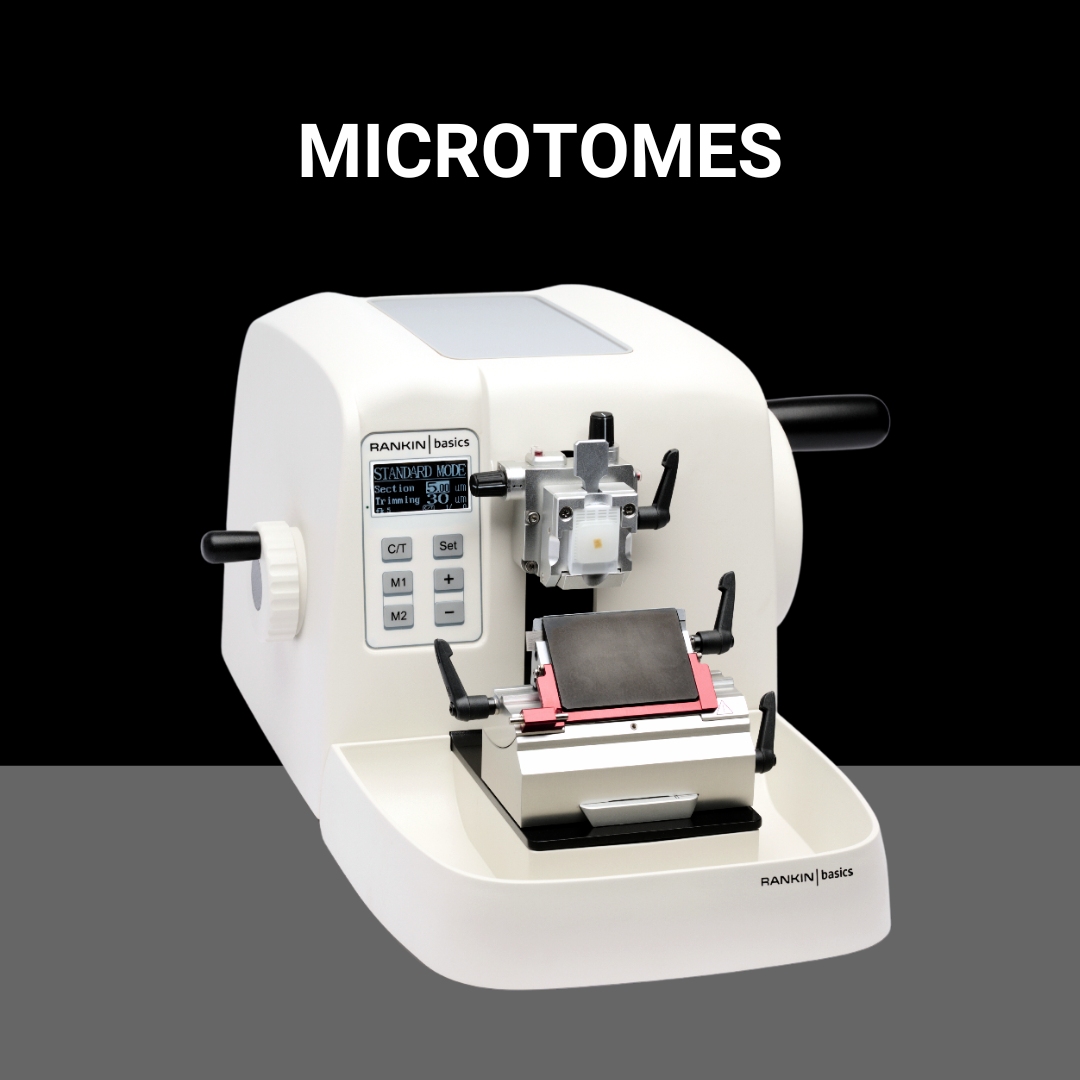
Shop All BrandsNew and refurbished equipment for your histology lab. Tissue processors, cassette & slide printers, embedding stations, microtomes, cryostats, slide stainers, and coverslippers.
Shop NowMicroscope slides, tissue cassettes, coverslips, paraffin, microtome blades, control slides, and more.
Shop NowNew and used repair parts for the most popular histology equipment.
Shop NowABOUT
We supply a comprehensive line of new and refurbished histology lab equipment, supplies, consumables, repair parts, accessories, instrument PM and repair service, and service agreements.
Accordion content.
Accordion content.
Accordion content.

Recent Articles
- Histology Educational Series – Part 1: Slide Labeling
 Every histology laboratory must have a slide labeling system. If you work in a small laboratory, this system may be as simple as using a solvent-resistant pen to hand-write your slides. Larger laboratories have moved forward to incorporate bar-code tracking systems into their LIS that can integrate with a slide printing module. Studies have shown that …
Every histology laboratory must have a slide labeling system. If you work in a small laboratory, this system may be as simple as using a solvent-resistant pen to hand-write your slides. Larger laboratories have moved forward to incorporate bar-code tracking systems into their LIS that can integrate with a slide printing module. Studies have shown that … - Rankin Biomedical moves headquarters to 35,000 square-foot facility
 This past January, Rankin Biomedical, a family-owned histology laboratory supply company, moved to a new facility with a significantly larger warehouse space. The larger facility has undoubtedly increased workflow and productivity along with allowing the company to store more inventory and efficiently handle growing customer orders. Since the move, Rankin Biomedical has been able to …
This past January, Rankin Biomedical, a family-owned histology laboratory supply company, moved to a new facility with a significantly larger warehouse space. The larger facility has undoubtedly increased workflow and productivity along with allowing the company to store more inventory and efficiently handle growing customer orders. Since the move, Rankin Biomedical has been able to … - The Anti-Roll Plate Issue: Preventing Section Bunching
 I can never get sections to slide under the anti-roll plate on the cryostat. The sections always seem to bunch up on the edge of the anti-roll plate. What do I need to do to get it to work right? The anti-roll plate is a square or rectangle of plastic or glass that rests against …
I can never get sections to slide under the anti-roll plate on the cryostat. The sections always seem to bunch up on the edge of the anti-roll plate. What do I need to do to get it to work right? The anti-roll plate is a square or rectangle of plastic or glass that rests against …


